MO teorie a konjugované vazby pí
Výhoda použití MO teorie pro pochopení vazeb v organických molekulách se stává zřejmější, když uvažujeme o vazbách pí. Uvažujme nejprve o vazbě pí v ethenu z hlediska teorie MO (v tomto příkladu nebudeme brát v úvahu vazby s v molekule a budeme uvažovat pouze o vazbě π). Začneme dvěma atomovými orbitaly: jedním nehybridizovaným 2p orbitalem od každého uhlíku. Každý z nich obsahuje jeden elektron. V teorii MO se tyto dva atomové matematicky spojí a vytvoří dva molekulové orbitaly pi, jeden nízkoenergetický vazebný orbital pi a jeden vysokoenergetický antivazebný orbital pi*.
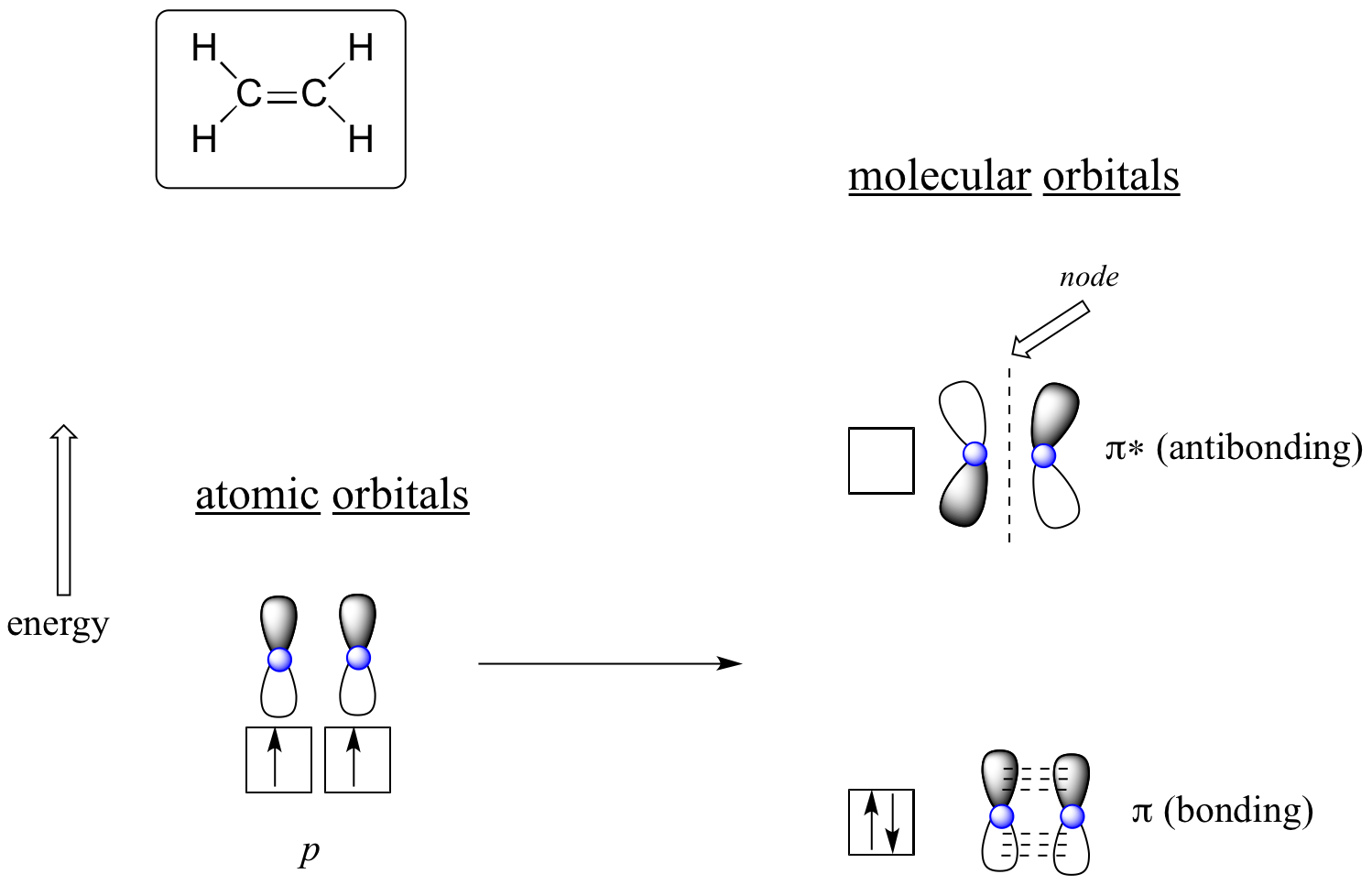
Molekulární orbitaly pro ethen (ethylen)
Ve vazebném orbitalu pi spolu oba zastíněné laloky orbitalů p konstruktivně interagují, stejně jako dva nezastíněné laloky (nezapomeňte, že libovolná volba zastínění představuje matematická znaménka (+) a (-) pro matematickou vlnovou funkci popisující orbital). V molekulovém orbitalu dochází ke zvýšené elektronové hustotě mezi oběma jádry uhlíku – jedná se o vazebnou interakci.
V antivazebném pi* orbitalu s vyšší energií interaguje zastíněný lalok jednoho p orbitalu destruktivně s nezastíněným lalokem druhého p orbitalu, což vede k uzlu mezi oběma jádry a celkovému odpuzování mezi jádry uhlíku.
Znovu použijeme princip „budování“ a umístíme dva elektrony do vazebného molekulového orbitalu pi s nižší energií. Antivazebný orbital pi* zůstává prázdný.
Příště se budeme zabývat molekulou 1,3-butadienu. Pouze z teorie valenčních orbitalů bychom mohli očekávat, že vazba C2-C3 v této molekule, protože se jedná o vazbu sigma, bude moci volně rotovat.

Experimentálně však bylo zjištěno, že existuje významná překážka rotace kolem vazby C2-C3 a že celá molekula je planární. Kromě toho je vazba C2-C3 dlouhá 148 pm, což je kratší než typická jednoduchá vazba uhlík-uhlík (asi 154 pm), i když delší než typická dvojná vazba (asi 134 pm).
Molekulární orbitální teorie tato pozorování vysvětluje pomocí konceptu delokalizovaných vazeb pí. V tomto obraze se čtyři atomové orbitaly 2p matematicky spojují do čtyř molekulových orbitalů pi s rostoucí energií. Dva z nich – vazebné orbitaly pi – mají nižší energii než atomové orbitaly p, z nichž jsou vytvořeny, zatímco dva z nich – antivazebné orbitaly pi* – mají vyšší energii.

Molekulární orbital pi1 s nejnižší energií má pouze konstruktivní interakci a nulové uzly. Energeticky vyšší, ale stále nižší než izolované p orbitaly, je orbital pi2, který má jeden uzel, ale dvě konstruktivní interakce – celkově se tedy stále jedná o vazebný orbital. Podíváme-li se na dva antivazebné orbitaly, pi3* má dva uzly a jednu konstruktivní interakci, zatímco pi4* má tři uzly a nulovou konstruktivní interakci.
Podle aufbauova principu jsou čtyři elektrony z izolovaných atomových orbitalů 2pz umístěny ve vazebných MO pi1 a pi2. Protože pi1 zahrnuje konstruktivní interakci mezi C2 a C3, existuje v molekule 1,3-butadienu určitý stupeň vazebné interakce pi mezi těmito dvěma uhlíky, což vysvětluje její kratší délku a bariéru rotace. Obrázek valenční vazby 1,3-butadienu ukazuje obě vazby pí jako vzájemně izolované, přičemž každý pár elektronů pí „uvízl“ ve své vlastní vazbě pí. Teorie molekulových orbitalů však předpovídá (přesně), že čtyři pi elektrony jsou do jisté míry delokalizovány neboli „rozprostřeny“ po celém systému vazeb pí.
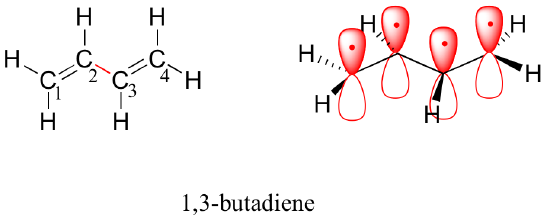
pohled vyplňující prostor
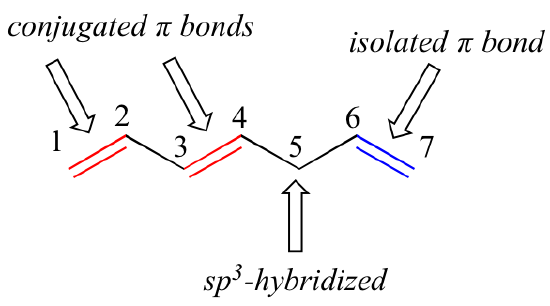
1,3-butadien je nejjednodušším příkladem systému konjugovaných vazeb pí. Aby mohly být dvě nebo více vazeb pi považovány za konjugované, musí být od sebe odděleny pouze jednou jedinou vazbou – jinými slovy, nesmí zde být intervenující sp3-hybridizovaný uhlík, protože ten by rozbil překrývající se systém paralelních p orbitalů. Například v níže uvedené sloučenině jsou dvojné vazby C1-C2 a C3-C4 konjugované, zatímco dvojná vazba C6-C7 je od ostatních dvou vazeb pi izolována sp3-hybridizovaným C5.
Velmi důležitý koncept, který je třeba mít na paměti, je, že s konjugací je spojena vlastní termodynamická stabilita. Tuto stabilitu lze experimentálně změřit porovnáním hydrogenačního tepla dvou různých dienů. (Hydrogenace je typ reakce, o kterém se dozvíme mnohem více v kapitole 15: v podstatě se jedná o proces přidání molekuly vodíku – dvou protonů a dvou elektronů – k vazbě p). Při „hydrogenaci“ dvou konjugovaných dvojných vazeb 1,3-pentadienu za vzniku pentanu se na jeden mol vzniklého pentanu uvolní přibližně 225 kJ. Porovnejte to s přibližně 250 kJ/mol, které se uvolní při hydrogenaci dvou izolovaných dvojných vazeb v 1,4-pentadienu za vzniku pentanu.
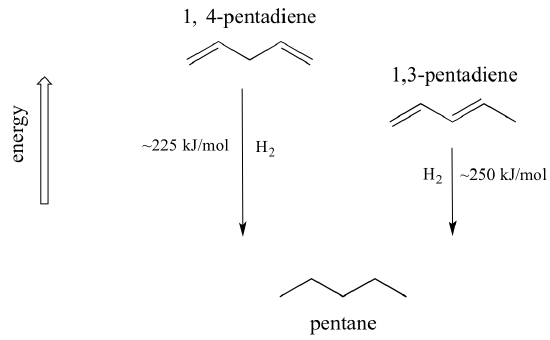
Konjugovaný dien má nižší energii: jinými slovy je stabilnější. Obecně jsou konjugované vazby pí stabilnější než izolované vazby pí.
Konjugované soustavy pí mohou zahrnovat kromě uhlíku i atomy kyslíku a dusíku. V metabolismu tukových molekul se některé z klíčových reakcí týkají alkenů, které jsou konjugovány s karbonylovými skupinami.

V kapitole 4 uvidíme, že teorie MO je velmi užitečná při vysvětlování, proč organické molekuly, které obsahují rozšířené systémy konjugovaných vazeb pí, mají často výrazné barvy. Beta-karoten, sloučenina zodpovědná za oranžovou barvu mrkve, má rozšířený systém 11 konjugovaných vazeb pí.
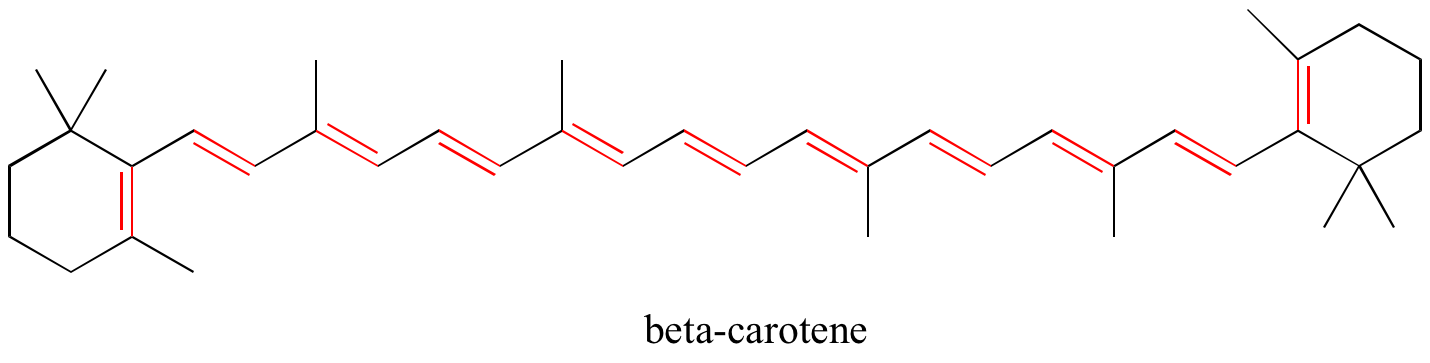
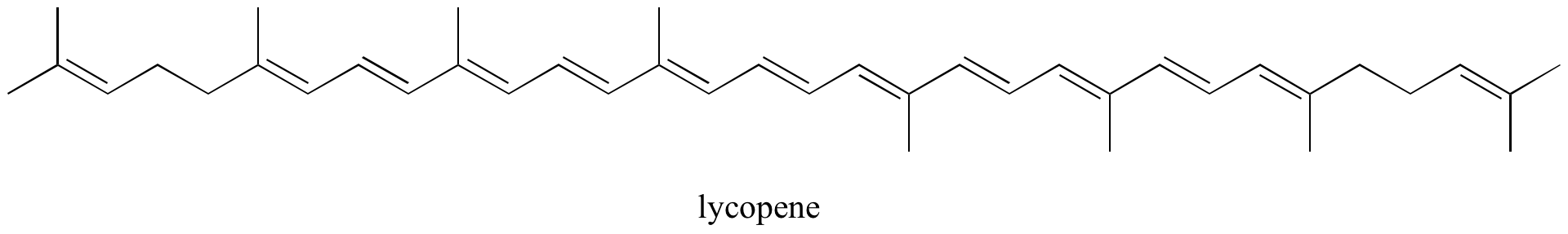
Cvičení: Určete všechny konjugované a izolované dvojné vazby v níže uvedených strukturách. Určete všechny izolované a konjugované vazby pí v lykopenu, červeně zbarvené sloučenině v rajčatech. Kolik elektronů pí je obsaženo v konjugovaném systému pí?
Řešení cvičení
Aromatičnost – konečná konjugovaná soustava
Teorie molekulových orbitalů je zvláště užitečná při vysvětlování jedinečných vlastností aromatických sloučenin, jako je benzen:
3D interaktivní model benzenu
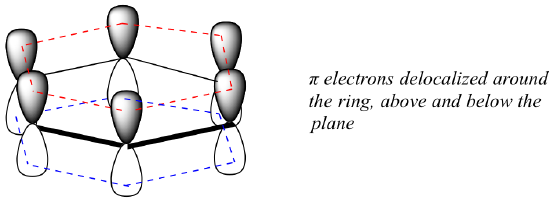
Ačkoli se benzen nejčastěji kreslí se třemi dvojnými a třemi jednoduchými vazbami, ve skutečnosti jsou všechny vazby uhlík-uhlík přesně stejně dlouhé (138 pm). Kromě toho jsou vazby pí v benzenu výrazně méně reaktivní než „normální“ vazby pí, ať už izolované, nebo konjugované. Něco ve struktuře benzenu způsobuje, že jeho uspořádání vazeb pí je obzvláště stabilní. Toto „něco“ má své jméno: nazývá se to „aromatičnost“.
Co přesně je touto „aromatickou“ vlastností, která činí vazby pí v benzenu tak stabilními? Odpověď na tuto otázku z velké části spočívá v tom, že benzen je cyklická molekula, v níž jsou všechny atomy kruhu sp2-hybridizovány. To umožňuje delokalizaci elektronů pí v molekulových orbitalech, které se rozprostírají po celém kruhu, nad i pod rovinou. Aby k tomu mohlo dojít, musí být samozřejmě kruh rovinný – jinak by se p orbitaly nemohly správně překrývat. Benzen je skutečně známý jako plochá molekula.
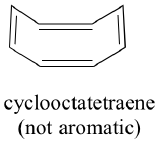
Mají všechny cyklické molekuly se střídajícími se jednoduchými a dvojnými vazbami stejnou aromatickou stabilitu? Odpověď ve skutečnosti zní „ne“. Níže zobrazený osmičlenný cyklooktatetraenový kruh není plochý a jeho π vazby reagují jako „normální“ alkeny.
Je zřejmé, že k aromatičnosti je potřeba něco víc, a to lze nejlépe vysvětlit pomocí teorie molekulových orbitalů. Podívejme se na energetický diagram molekulových orbitalů pi v benzenu.
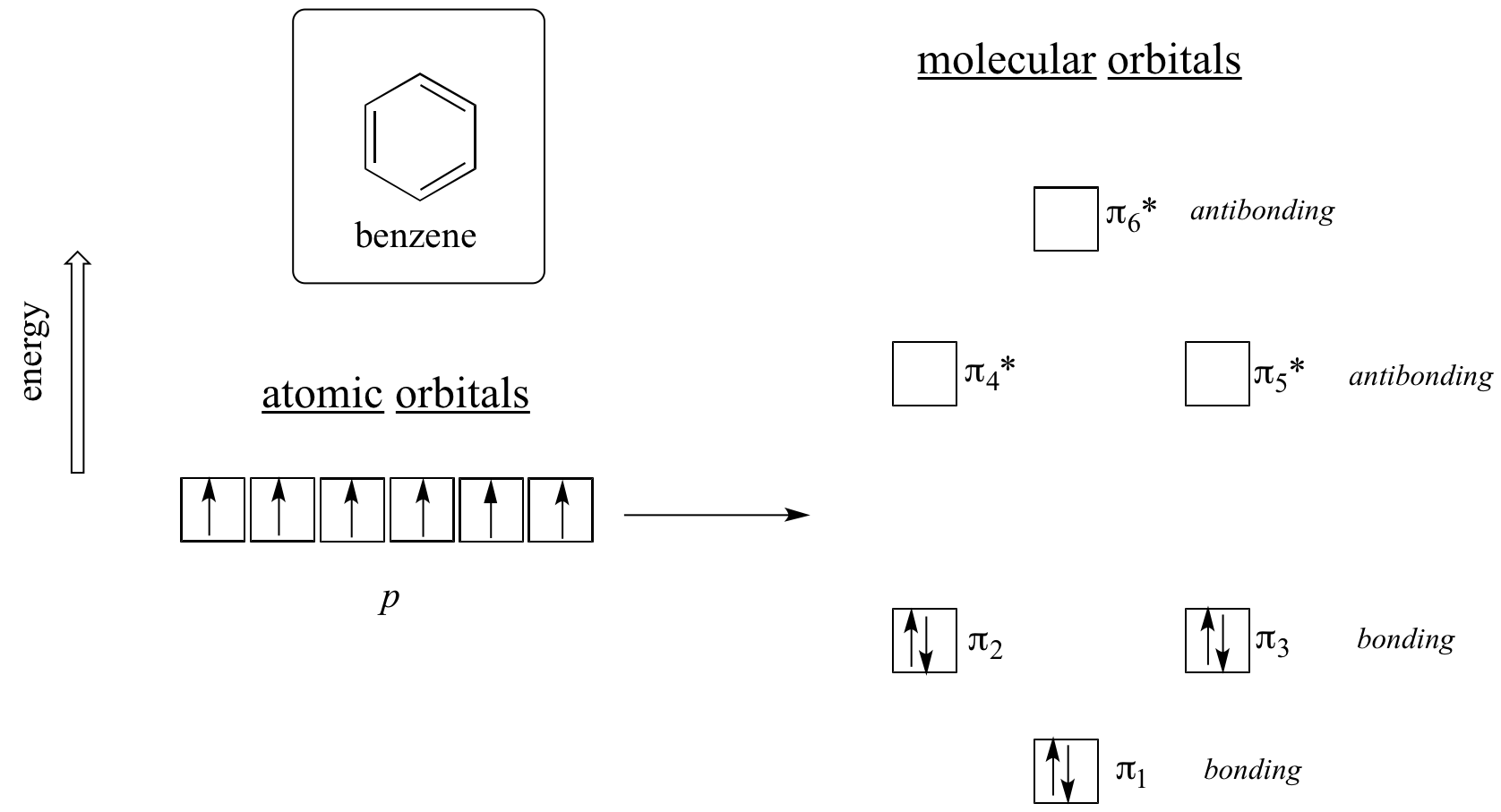
Kvantově mechanické výpočty nám říkají, že šest molekulových orbitalů pi v benzenu, vytvořených ze šesti atomových orbitalů p, zaujímá čtyři samostatné energetické hladiny. Pí1 a pí6* mají jedinečné energetické hladiny, zatímco páry pí2 – pí3 a pí4*- pí5* jsou degenerované, což znamená, že jsou na stejné energetické hladině. Když pomocí aufbauova principu zaplníme tyto orbitaly šesti elektrony pí v benzenu, zjistíme, že vazebné orbitaly jsou zcela zaplněny a antivazebné orbitaly jsou prázdné. To nám dává dobré vodítko ke zdroji zvláštní stability benzenu: plný soubor vazebných MO je v mnoha ohledech podobný „plné slupce“ elektronů v atomových orbitalech stabilních vzácných plynů helia, neonu a argonu.
Provedeme nyní totéž pro cyklooktatetraen, o kterém jsme se již dozvěděli, že není aromatický.
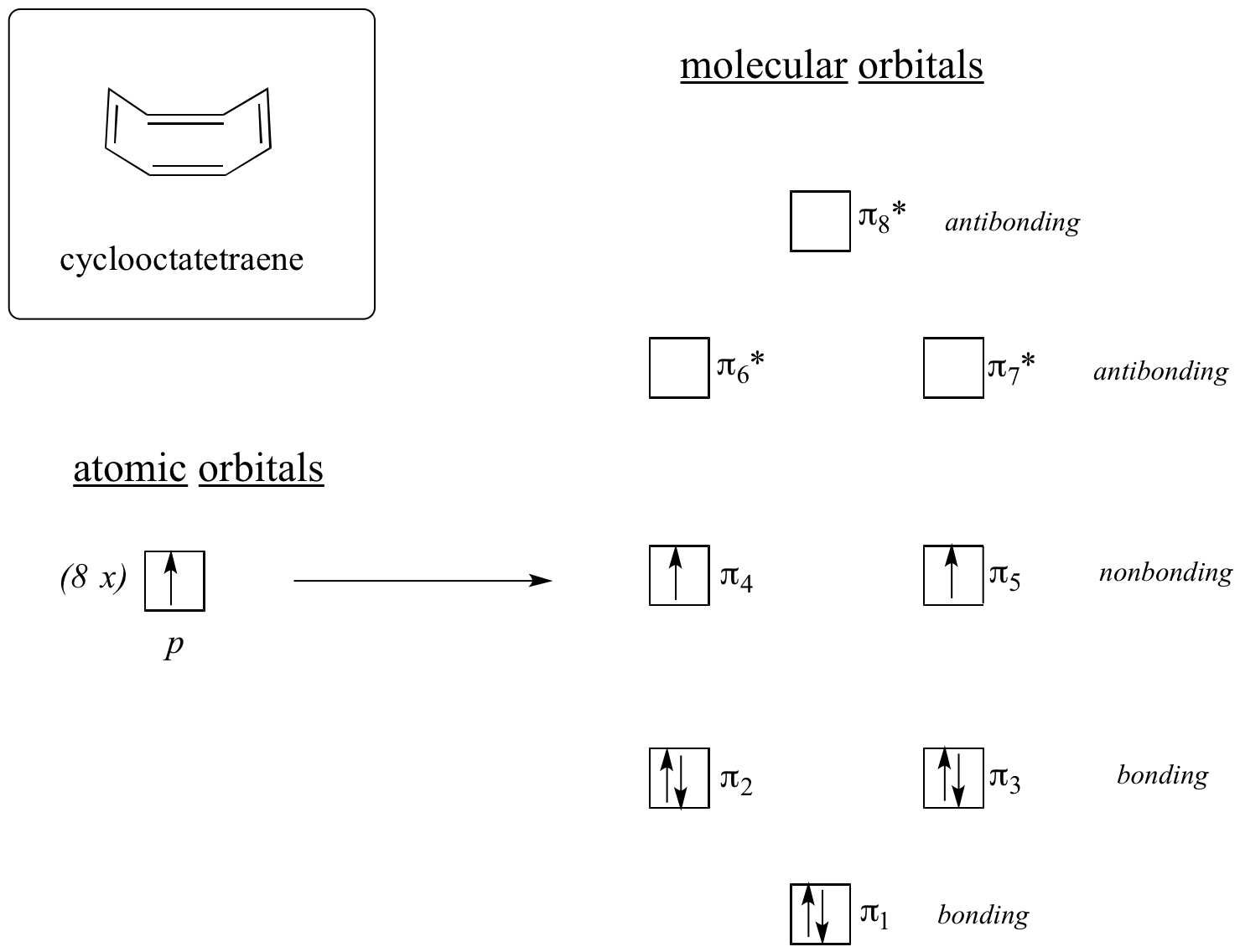
Výsledek výpočtů molekulových orbitalů nám říká, že MO s nejnižší a nejvyšší energií (pi1 a pi8*) mají jedinečné energetické hladiny, zatímco ostatních šest tvoří degenerované páry. Všimněte si, že pi4 a pi5 jsou na stejné energetické hladině jako izolované atomové orbitaly 2pz: nejsou tedy ani vazebné, ani antivazebné, spíše se označují jako nevazebné MO. Doplníme-li MOs osmi pi elektrony v molekule, zjistíme, že poslední dva elektrony jsou nepárové a spadají do dvou degenerovaných nevazebných orbitalů. Protože nemáme dokonale vyplněnou slupku vazebných MO, není naše molekula aromatická. V důsledku toho se každá z dvojných vazeb v cyklooktatetraenu chová spíše jako izolovaná dvojná vazba.
Prozatím je důležitým cílem výuky rozpoznat systémy konjugovaných vazeb pí a pochopit, že benzen je mimořádně stabilní a vykazuje vlastnost zvanou aromaticita. Aromatičnost a chemie aromatických sloučenin je poměrně složitá a podrobněji se jí budeme věnovat v dalších kapitolách tohoto textu.
Organická chemie s biologickým zaměřením autor: Tim Soderberg (University of Minnesota, Morris)